Why is Process Safety Important?
From synthesizing a new API to developing a novel polymer, chemical reactions and the products they form are front and center in the world in which we live. One of the biggest challenges facing those trying to bring these products to market is scale-up: taking small, lab-based synthesis through pilot plant to full-scale manufacture.
It is well-recognized that there are numerous safety risks involved in scaling up chemical processes. Central to these are the changes in heat-loss behavior, which differ with scale:
- Many reactions are exothermic. Due to the small scale, this heat can quickly dissipate to the surrounding environment in a lab-based environment. However, the reaction may require additional active temperature management on scale-up to ensure safe reaction conditions.
- Components in the reaction may become unstable under certain operating conditions, including storing large volumes, which can cause additional hazards that require management.
The risks associated with scale-up have potentially hazardous, even fatal, consequences if not addressed. Thus, thermal and pressure hazards must be considered, identified, assessed, and mitigated at each stage of product development. To achieve these objectives, a number of questions need to be answered, which will depend on the step of the scale-up process (Figure 1). By designing safety into the product development from the earliest stages – a Safety by Design, if you like – one can feel confident that the hazards associated with scale-up to manufacturing can be successfully addressed.

How to apply Process Safety into the Development Workflow?
Applying a safety-by-design approach within the development workflow has safety and cost benefits.
- Broadly speaking, process safety scale-up follows four main stages:
- Screening for potential hazards
- Evaluation of the main reactions, including possible unintended side reactions
- Evaluation of “what if” worst case scenarios and subsequently implementing mitigation and control strategies.
These steps naturally map onto the product development funnel. At the top of the funnel, the tests need to be simple and quick to address the number of samples. At this stage, the information provided can be quite limited. At the bottom of the funnel, poor candidates have been eliminated from consideration, and more information is required. Tests at this stage will be longer, and the experimental complexity will be greater than in discovery.
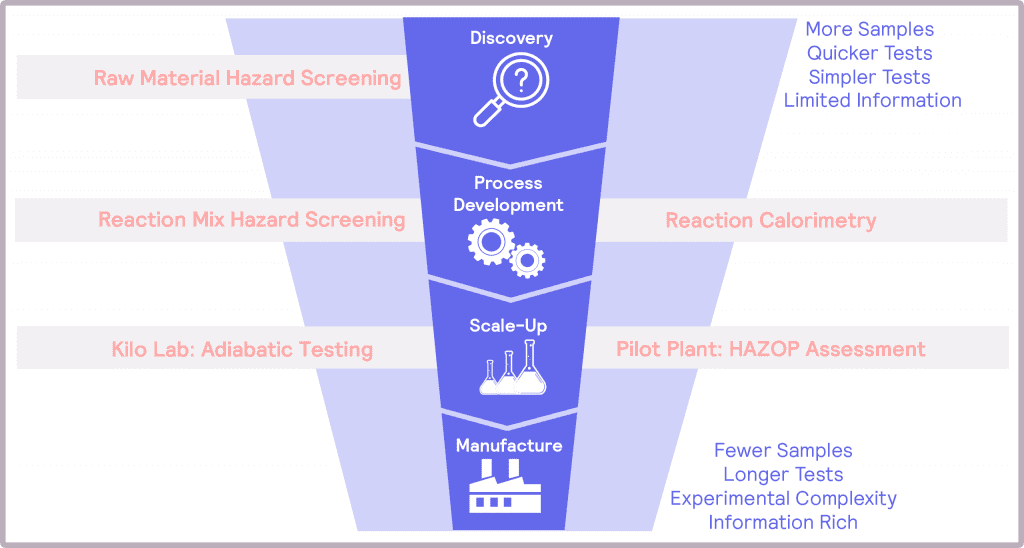
At the process development stage, the target molecule has been identified, and its synthetic route has been determined. The work at this stage is to develop and optimize the process with a view to manufacturing at scale. In terms of safety, the desired main reaction needs to be assessed and understood, alongside identifying potential additional thermal and pressure hazards as a result of the operating conditions. An example of these hazards could be undesired side products that are more unstable than the desired product.
During scale-up, the different operating conditions are evaluated to assess their impact on the hazards. This information is used to optimize the conditions for safety. Finally, with the operating conditions optimized and the potential hazards identified, it is time to assess the magnitude of these hazards prior to full scale-up.
H.E.L’s Process Safety Solutions
Consideration of process safety throughout the product development pipeline from discovery to scale-up is essential to ensure the safe scale-up of chemical processes.
In Process Development, it is vital to understand how hazardous the selected synthesis reaction is and whether the plant cooling capacity is sufficient to mitigate these potential hazards. Hazard screening and reaction calorimetry both have a role to play when understanding a reaction itself. The move to scale-up is characterized by an increasing focus on the true extent of identified hazards, so plant safety design can be optimized accordingly. Adiabatic testing and HAZOP (hazard and operability) assessments come to the fore here. These considerations are summarized in the product development funnel in Figure 3.
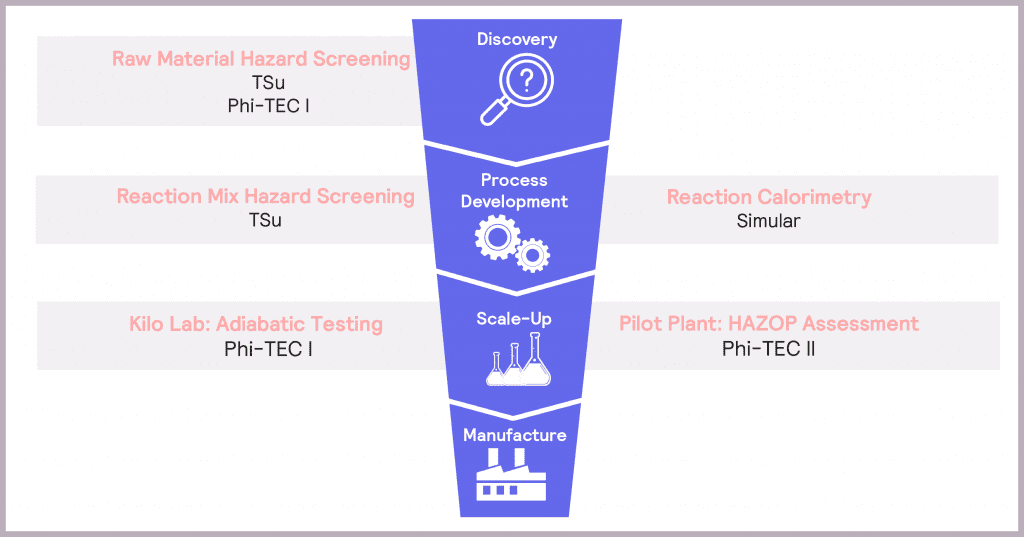
Making safety an integral component of product development – by using a variety of tools to identify, assess and then mitigate the potential hazards at each stage of the process – ensures confidence in delivering a successful and efficient route to market.

TSu | Thermal and Pressure Hazard Screening Platform
Home › Products › Process Safety › TSuTSuFully customisable thermal ...

Phi-TEC I | Bench-top, adiabatic calorimeter
Home › Products › Process Safety › Phi-TEC IPhi-TEC IFully customisa...

Phi-TEC II | Bench-top, low phi-factor, adiabatic calorimeter
Home › Products › Process Safety › Phi-TEC IIPhi-TEC IIFully customi...

Simular | Process Development Reaction Calorimeter
Home › Products › Process Safety › SimularSimularFully customisable ...
Discovery
How does early raw material hazard screening speed up the decision process?
Rapid hazard screening of raw materials at the front end of development allows earlier decisions to be made on how to develop the desired reaction and the synthetic route.
It is vital to know early in product development if the raw materials are likely to have thermal stability issues, such as the production of non-condensable gases, which could pose an explosion risk on scale-up.
Key questions to answer are:
- What is the thermal decomposition profile of the raw material? In particular, does the decomposition of the raw material start to increase significantly at a ‘low’ temperature (onset decomposition temperature)? If the desired synthesis reaction reaches this onset temperature via an initial exothermic event, this could cause the reaction to increase further in temperature, creating a secondary thermal runaway.
- Is there a rapid rate of pressure change during the exothermic event? This would indicate that the reactant could pose an over-pressurization hazard during a thermal runaway.
Micro-scale calorimetry is typically used to perform a quick screening assessment on the thermal properties of the raw material, with Differential Scanning Calorimetry (DSC) being the typical method. DSC is a thermoanalytical technique in which the difference in the amount of heat required to increase the temperature of a sample and reference is measured as a function of temperature. However, this method does not provide critical information on pressure change, which is vital for assessing the over-pressurization hazard. It also suffers from the challenge that representative samples cannot always be obtained. For raw, pure materials, this is less of an issue, particularly when one only needs an initial assessment on whether to proceed further with the material. However, it becomes more significant as one moves to screening reaction mixes and further into process development, and rapid screening of larger sample sizes is required. This is especially true for poorly mixed samples and slurries.
Solutions
The TSu (thermal screening unit) is designed to record both pressure and temperature data allowing an in-depth characterization of the chemical reaction of interest. Both temperature and pressure can be dangerous in industrial processes since hot equipment can result in burns and injuries, but over-pressurized equipment can cause deadly explosions. Fundamental parameters, such as the onset temperature of decomposition (Td) and the rate of pressure increase, can be easily determined, enabling the hazard assessment of the material used and providing critical information for process safety. It is designed to be compact and easy to use but allows the user a larger sample mass and, consequently, a more accurate model for production scale. Its wide range of temperatures means that it can easily determine the onset temperature of decomposition. TSu’s rapid sample turn-around helps to make it a cost-effective unit, reducing the running cost.
TSu | Thermal and Pressure Hazard Screening Platform
Home › Products › Process Safety › TSuTSuFully customisable thermal ...
Process Development
Understanding the desired reaction
Here, the key pieces of information to extract are:
- What is the energy evolved in the reaction?
- What is the cooling capacity required in the manufacturing plant to maintain an isothermal reaction?
- What is the subsequent rise in temperature when no cooling is applied?
Chemical reactions involve the transformation of reactants into products; heat will be exchanged with the environment in the process. In the case of exothermic reactions, energy will be released, and if the reaction is endothermic, energy will be absorbed. Understanding the thermodynamic nature of this transformation is fundamental. If endothermic and external energy is not supplied, the progression of the reaction can be very slow, making it inefficient. But on the other hand, if a chemical process is exothermic, associated risks might come, such as high temperature or high pressures.
Calorimeters can be used to perform the characterization of the chemical reaction. If the temperature is to be kept constant in the reaction (isothermal conditions), energy needs to be supplied to maintain this condition. This would tell us how much energy the reaction produces or takes. These energy values scale linearly, so we can calculate the energy required for the industrial scale using the value obtained from the calorimeter in a small-scale experiment.
As an example, we can use the esterification of acetic anhydride with methanol, which produces ethyl acetate and acetic acid:
(CH3CO)2O + CH3OH → CH3COOCH3 + CH3COOH
This is an exothermic reaction that releases 66 kJ mol-1. A calorimeter, like a Simular can be used. In this case, we can use a semi-batch regime, adding the reactant (acetic anhydride) at a feed rate of 2 g·min-1 over 50 mins and keeping the temperature at 30°C. As the reaction progresses, heat is released, and the cooling system activates to maintain a constant temperature. The figure below shows the power supplied to the reactor to keep the reaction isothermal (in W), how much acetic anhydride has been supplied (in g), and the energy released (in J).
In the following example of a semi-batch esterification reaction process, acetic anhydride was added at a 2 g/minute dosing rate over 50 minutes to methanol in a 1 L reactor in the Simular reaction calorimeter. At the same time, the temperature was held isothermally at 30°C [1]. Figure 5 shows the:
- Reaction power (this is essentially the rate of heat generation) [pink trace]
- Energy released [blue trace]
- Feed rate [yellow trace]
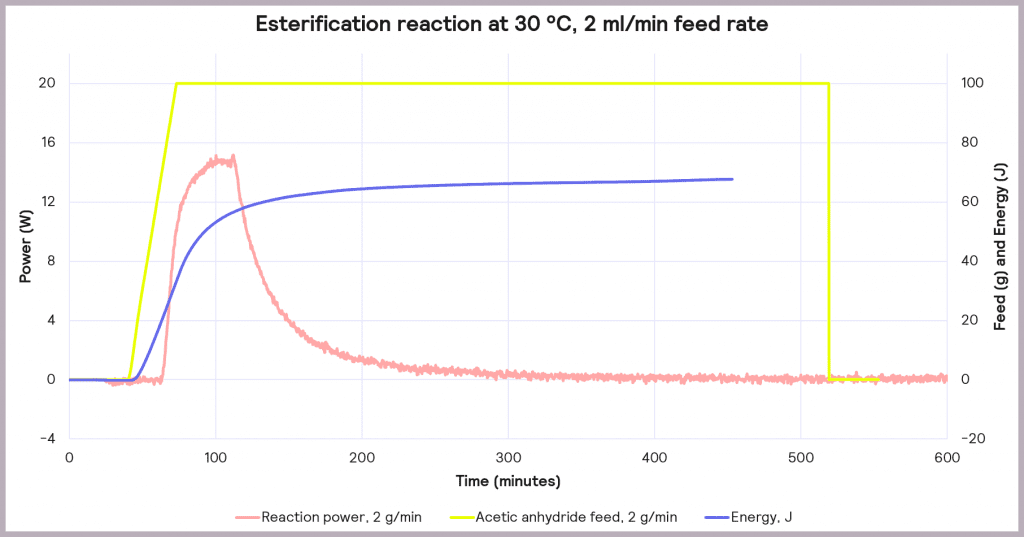
Under these conditions, it can be seen that the reaction power reaches a maximum of 15W. This means a cooling capacity of at least 15 W is required to keep this reaction isothermal. As a first check, these values can be proportionately scaled up: for example, the reaction was performed in a 1 L reactor, so for a 2000 L pilot reactor, this would indicate that at least a 30 kW (30,000 W) cooling capacity is required for the larger system.
Also, the measured value for the energy released during the reaction is around 66 J, which progresses even when the reactor feed has finished. The energy released after the feed stopped is 34.6 kJ. This allows us to know the maximum temperature of synthesis reactions using the following equation:
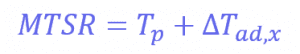
Where Tp is the temperature of the reactor (in this case, 30°C) and ∆𝑇𝑎𝑑,𝑥 is the adiabatic temperature increase. This is the change in the temperature when there is no heat exchange with the surroundings – so it just comes from the reaction itself. These values can be calculated using another equation:
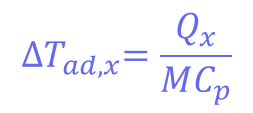
Where mCp is the thermal mass of the components (the mass multiplied by the heat capacity), and its value is 326 J°C-1. This means that the adiabatic temperature increase in the reactor is 106°C, so the total MTSR is 136°C. ∆𝑇𝑎𝑑,𝑥
Thus, reaction calorimetry enables the heat evolved during the desired reaction to be measured, which is then used to calculate the cooling capacity required to keep the system at the desired reaction temperature – Tp.
Solutions
The Simular measures the energy evolved in the reaction. Subsequently, this enables you to calculate the plant cooling capacity required to keep the reaction isothermal (Tp) and the maximum temperature the reaction can reach, helping predict the risks and reducing the hazards. For this reason, the Simular can be used to explore and design safer reaction conditions, thereby facilitating the optimization of safe operations and minimizing process risk.
Simular | Process Development Reaction Calorimeter
Home › Products › Process Safety › SimularSimularFully customisable ...
Identifying and understanding decomposition and secondary thermal runaway (the undesired reactions)
The use of calorimetry can help us characterize our target chemical reactions: power required to maintain isothermal conditions, runway reactions when cooling is not supplied, and what maximum temperature the reactor can reach.
However, additional reactions may occur in many cases when some of the reagents or products can undergo subsequent chemical reactions. The velocity of these side reactions can exponentially increase when the temperature rises due to the progression of the desired reactions. Among these side reactions, decomposition reactions are of special relevance. Decomposition reactions are those chemical reactions in which one reactant breaks down into two or more products.
A secondary thermal runaway is likely to occur if MTSR is greater than the onset temperature of a component in the reaction mix (Td). The onset temperature can be defined as the temperature at which the decomposition reaction starts to be detected. This scenario is illustrated in Figure 6.
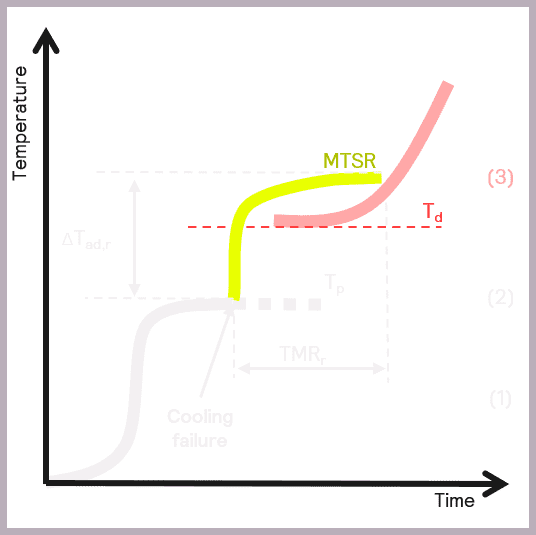
As previously described, MTSR is estimated from reaction calorimetry, while the onset temperatures of materials are determined using thermal screening tools, such as the Thermal Screening Unit (TSu).
A more accurate value of the onset temperature can be determined using adiabatic calorimetry. Still, it is often considered sufficient to use a more straightforward thermal screening tool at this stage. The TSu enables representative samples of the reaction mix to be screened and reliable onset temperatures determined for the reaction intermediates and products. Thermal stability studies also need to take place on the reaction waste stream, which the TSu also facilitates with isothermal stability screening. Just as it is important to identify hazards during the reaction itself, it is also important to understand if there are any longer-term stability issues in the waste products formed so that these issues can also be mitigated.
Suppose it becomes apparent that there is a risk of a secondary thermal runaway occurring. In that case, it is necessary to determine the adiabatic temperature rise of the decomposition/secondary reaction. This allows an assessment to be made as to whether there will be a sufficient emergency cooling capacity to deal with the increase in temperature.
As this reaction involves a secondary thermal runaway, scaling up is generally a hazardous process. Therefore, although these parameters can be estimated from micro-calorimetry, it is advisable to use adiabatic calorimetry to get an accurate assessment.
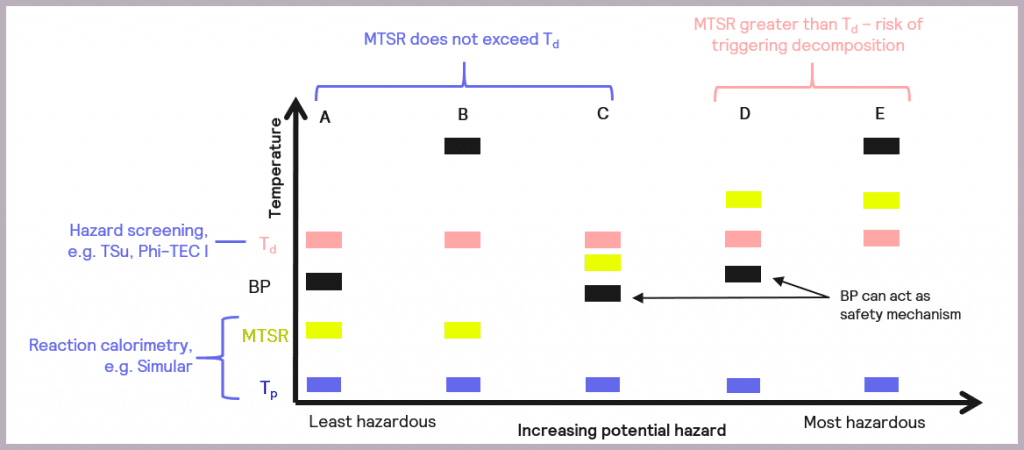
The data collected enables the criticality of the reaction to be classified, as described by Stoessel [2], as a possible assessment of hazards and depicted in Figure 7.
The schematic references the key reaction parameters introduced in Figure 5:
- the isothermal temperature of the reaction (Tp)
- the decomposition temperature of a component in the reaction mix (Td)
- the Maximum Temperature of the Synthesis Reaction (MTSR)
It also introduces an additional parameter, the boiling point (BP) of the solvent or active chemicals used, as this is another factor that needs to be taken into account when considering the overall safety of a reaction. The relative order of these parameters in terms of temperature determines the criticality of the reaction.
- Scenario (A): the thermal order of the parameters is Tp<MTSR<BP<Td. This represents the least hazardous reaction scenario, and no special measures are required as the MTSR does not exceed that of the BP of the solvent nor the Td.
- Scenario (B): as with Scenario (A), the MTSR does not exceed Td, although in this case, the BP of the solvent is higher than Td (Tp<MTSR< Td< BP). This means that, unlike Scenario (A), the BP will not act as a limit on the maximum temperature the reaction will attain, and heat accumulation conditions should be avoided.
- Scenario (C): the MTSR is higher than the BP but less than Td (Tp< BP< MTSR<Td). This means that risk reduction measures are required but that the BP of the solvent could be used as a non-ideal safety mechanism for the reaction.
- Scenario (D): the MTSR is higher than Td, which means there is a risk that the decomposition could be triggered should the system fail. However, the BP of the solvent could be used as a limiting mechanism to prevent triggering the decomposition reaction, as BP is lower than Td (Tp<BP<Td<MTSR). In this case, risk reduction measures are required.
- Scenario (E): as with Scenario (D), the MTSR is greater than Td, which means there is a risk that the decomposition could be triggered. However, the BP of the solvent is also above that of Td, so it can’t be used as a safety barrier (Tp<Td<MTSR<BP). In this case, emergency control measures are also required, which may include redesigning the process.
Although the Stoessel Diagram can help assess how hazardous a reaction is, caution needs to be exercised [3]. For example, the solvent boiling point temperature assumes atmospheric pressure. Therefore, if the operating pressure becomes higher than this after a cooling failure, then the solvent boiling temperature also increases, effectively removing the safety mechanism in Scenario C and Scenario D. Similarly, if the solvent is rapidly vaporized by a fast energy release from decomposition, the reactor could become over-pressurized before the plant pressure relief mechanisms kick in. Therefore, while the Stoessel Diagram is useful to facilitate the development of intrinsically safer processes, it is important to recognize that a wider range of potential hazards must also be considered, pressure chief among them.
Solutions
Suppose the MTSR is greater than a component’s onset temperature (Td) within the reaction mix. In that case, an undesired side reaction or decomposition may be triggered, leading to a secondary thermal runaway.
To better understand the risks of thermal runaway in a reaction, the TSu (Thermal Screening Unit) can be used. The TSu supports large-volume measurements, enabling representative samples of the reaction mix to be screened and reliable onset temperatures to be determined for the reactants, intermediates, and products in the reaction mix. It also enables the vital assessment of pressure events in the reaction mix and can find use in studying the waste streams from reactions.
TSu | Thermal and Pressure Hazard Screening Platform
Home › Products › Process Safety › TSuTSuFully customisable thermal ...
How Can The Operating Conditions Be Modified To Reduce The Risks Identified?
Stoessel diagrams can help us understand how risky chemical processes are, including desired reactions and secondary reactions (such as decomposition reactions). Stoessel classification identifies 5 possible scenarios, ranked from least dangerous to most dangerous. The commonality that the three first categories have, is that the maximum temperature of synthesis reaction (MTSR – the maximum temperature the reactor would reach if a failure in cooling occurs) stays under the temperature of decomposition (Td, temperature at which some of the reactants or products start to break down at a detectable rate).In the case of scenarios (D) & (E), it is necessary to consider if the MTSR can be reduced by altering the conditions of the reaction to reduce the levels of accumulated reagent.
There are several ways this could be addressed, such as [1]:
- Increasing the temperature of the reaction
- Increasing the pressure of the reaction
- Increasing the agitation of the reaction mix
- Adding a catalyst
- Reducing the feed rate of the reactant
The first four methods will increase the reaction rate and thus increase the reagent’s consumption rate. However, it must be balanced against the subsequent increase in the rate of heat evolved, and so an assessment on whether the plant’s cooling capacity can contain this increase in heat.
Reducing the reactants’ feed rate will reduce the reagent accumulation rate as it effectively allows more time for the reaction to occur and the rate-limiting reagent to be consumed before it accumulates to hazardous levels.
Exploring the impact of varying these different factors on the reaction is desirable. The Simular enables users to experiment in this way by creating experiments that facilitate the identification of optimum operating conditions for reducing thermal risk. For example, an esterification reaction in which acetic anhydride is fed into a reactor containing methanol can be kept at different temperatures. So let us analyze how different feed rates can affect the power needed to keep the conditions isothermal and accumulated reagent.
Reducing The Feed Rate
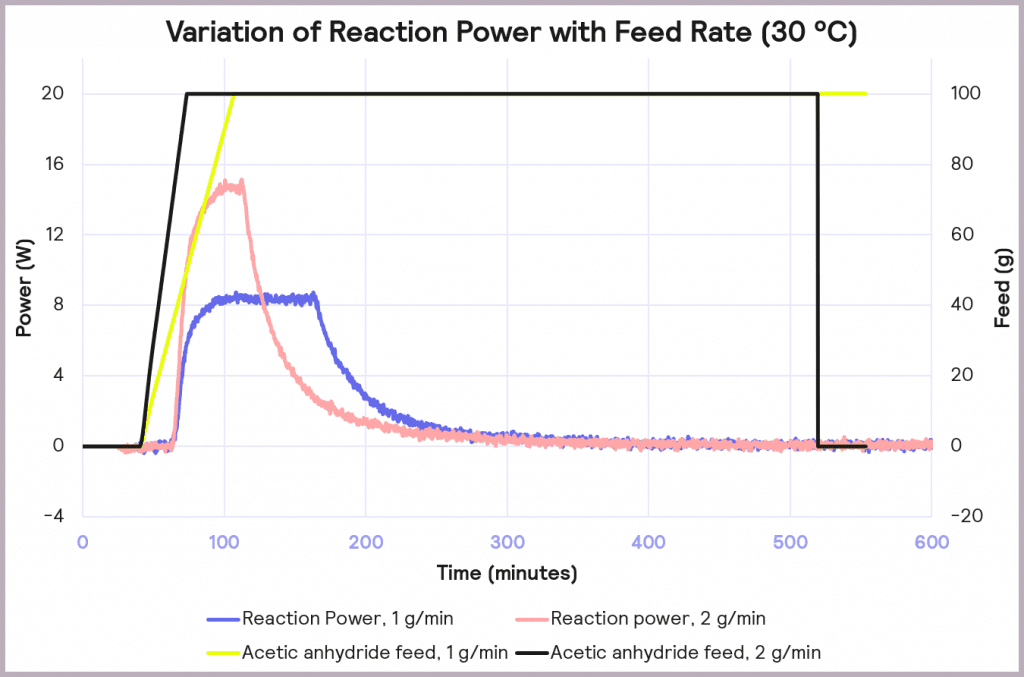
The effect of reducing the feed rate from 2 g/min to 1 g/min, while keeping the temperature of the process at 30 ⁰C, is illustrated in Figure 8. It reduces the accumulation of reagent to 32% at the point at which the feed stops, and it reduces the maximum reaction power from 15 W to 8 W. This reduction in maximum reaction power significantly reduces the cooling capacity required to keep the reaction isothermal and under thermal control.
Increasing the Temperature
The accumulated reagent level can be further reduced by increasing the temperature from 30 ⁰C to 40 ⁰C while maintaining the slower feed rate of 1 g/min. The impact of this is depicted in Figure 11. This reduces the level of reagent accumulated to 22% at the point at which the feed is stopped, but it does increase the maximum reaction power from 8 W to 10 W.
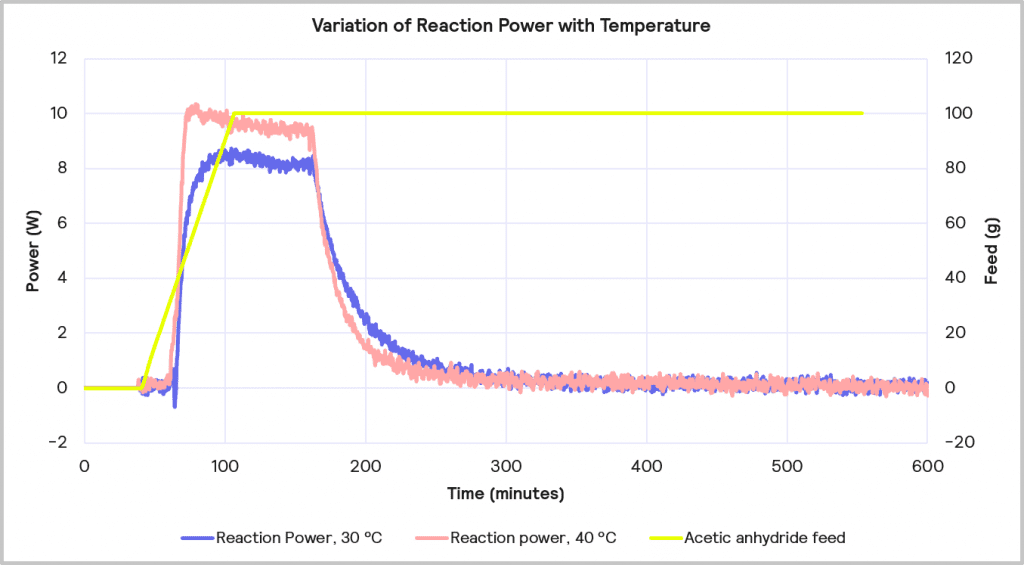
Cooling Capacity Considerations
Increasing the reaction temperature poses an interesting question: while the increase in temperature will result in less accumulated reagent and thus a lower MTSR, should cooling fail, and a thermal runaway occurs, it does require a larger cooling capacity to deal with the faster rate of heat evolved. As before, if it is assumed that the 1 L lab-based reactor is initially scaling up to a 2000 L reactor, the 10W reaction power for the reaction run at 40⁰C equates to a 20 kW cooling capacity requirement on scale-up versus the 16 kW required for the same reaction run at 30⁰C. Therefore, although the 1g/min feed rate with 40⁰C process temperature is the desired combination from a safety point of view in terms of reagent accumulation, it may be that a lower temperature is required if the cooling capacity of the pilot plant is <20 kW.
Another vital consideration is the efficiency of the cooling. As the scale of the reaction increases, the thermal transfer efficiency from the temperature-controlling system will likely decrease. Because of this, the cooling capacity will probably have to increase. A suitable safety factor must also be applied to the cooling capacity to ensure the system can handle unexpected thermal events.
Ideally, a process should be as close to a dose-controlled reaction as possible. In this scenario, the reactant is consumed as rapidly as it is added. Therefore, in the event of a cooling or agitation failure, it is usually sufficient to switch off the feed to prevent a thermal runaway.
However, in cases where accumulation can’t be avoided, it is necessary to undertake a more detailed analysis of the risk.
Solutions
The Simular measures the energy evolved in the reaction. Subsequently, this enables you to calculate the plant cooling capacity required to keep the reaction isothermal (Tp). It also allows for the Maximum Temperature of Synthesis Reaction (MTSR) to be calculated from the reaction data. For this reason, the Simular can be used to explore and design safer reaction conditions, thereby facilitating the optimization of safe operations and minimizing process risk.
Simular | Process Development Reaction Calorimeter
Home › Products › Process Safety › SimularSimularFully customisable ...
Scale-Up
Why Adiabatic Calorimetry?
Large-scale reactors, like those found in manufacturing and pilot plants, behave pseudo-adiabatically – i.e., very little heat is lost to the surroundings. This contrasts with the smaller vessels used in laboratories, where heat loss is much more significant. This poses a hazard on scale-up, as heat generated from a reaction will be retained within the system – at best, requiring more plant cooling and, at worst, triggering a thermal runaway. This is due to the decrease in surface area to volume ratio in larger vessels, which provides less area for heat dispersal.
Adiabatic calorimeters mimic these large-scale vessels in small-scale systems. An adiabatic calorimeter will eliminate increasing heat losses with the application of carefully modeled and controlled system temperature increases.
Broadly speaking, there are two types of adiabatic calorimeters that find use at different stages of scale-up. The more common, classic acceleration rate calorimetry or “ARC”-style adiabatic calorimeter, such as the Phi-TEC I, can be used earlier in the scale-up process as an additional reaction screening tool. The test cells used normally have a larger thermal mass than the reactants they contain and are described as having a high Phi (Φ) factor. A Phi factor is defined as [1]:
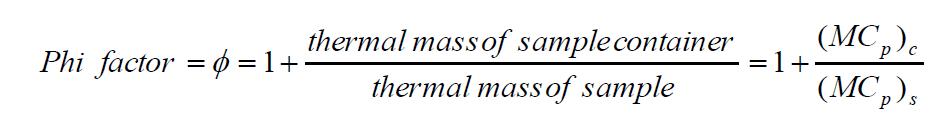
As a result, the data from these high Phi-factor test cells must be recalculated before the results can be scaled up.
However, the second type, such as the Phi-TEC II, uses low thermal mass test cells. This allows the Phi-TEC II to more closely simulate plant-scale adiabatic runaways and generate data that can be used to define the safety measures that will need to be put in place on a manufacturing scale. As such, it tends to be used at the final stages of scale-up, when all other safety bases have been explored, but the only possible route is to allow the runaway to progress.
The classic “ARC”-style adiabatic calorimeter sits between the basic screening tools used earlier in process development and the advanced, low Phi-factor adiabatic calorimeter used in the final scale-up. Accordingly, experiments undertaken with the Phi-TEC I deliver additional information on thermal hazards to that obtained from the TSu. Likewise, the Phi-TEC II provides even richer process information. The Phi-TEC I is often used when the initial screening tests identify that a reaction is highly exothermic and/or carries a significant risk of over-pressurization. A more detailed characterization of the potential thermal runaways is required in these cases.
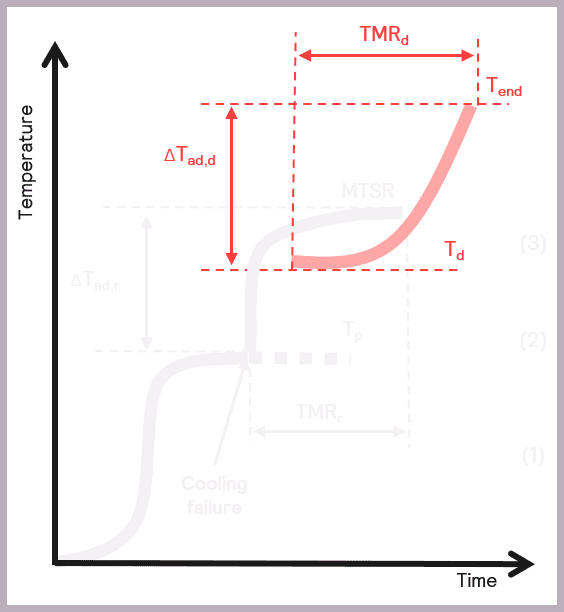
The Phi-TEC I allows the process safety chemist to carry out a detailed sample evaluation by studying the exotherm under adiabatic conditions, thus delivering an accurate determination of the:
- The onset temperature of any decompositions or side reactions (Td)
- The adiabatic temperature rise of any secondary thermal runaways (ΔTad,d)
- Time to Maximum Rate of the secondary thermal runaway (TMRd).
- The pressure increase during the decomposition or side reactions
Some of these parameters are illustrated schematically in Figure 10.
The Phi-TEC I ensures that exothermic events are fully tracked and pseudo-adiabatic conditions are maintained by providing a direct measurement of the sample temperature, coupled with a rapid guard heater response time to thermal changes,
However, it is important to realize that the rate of pressure change and maximum temperature determined are unlikely to represent anything that happens at the process scale, particularly if the reaction is gas-generating and volume becomes important. Representative values for these parameters can be obtained using low Phi factor test cells for adiabatic testing.
Solutions
When scaling up a process, accurate knowledge of an exothermic event is
vital to ensure the magnitude of the thermal runaway risk is fully understood. The Phi-TEC I provides a direct measurement of the sample temperature with a rapid response to thermal changes. It accurately tracks exothermic events and maintains the required adiabatic conditions.
Phi-TEC I | Bench-top, adiabatic calorimeter
Home › Products › Process Safety › Phi-TEC IPhi-TEC IFully customisa...
Phi-TEC II | Bench-top, low phi-factor, adiabatic calorimeter
Home › Products › Process Safety › Phi-TEC IIPhi-TEC IIFully customi...
From pilot plant to manufacture: Simulating plant conditions to inform safety control factors
Before the final scale-up to manufacturing, it is necessary to fully understand what safety control factors are required to mitigate the worst-case scenarios. The ability to fully simulate thermal runaway hazards under manufacturing conditions in the lab is a valuable way of understanding the process and ensuring the appropriate safety measures are planned and implemented prior to manufacturing.
Using an adiabatic calorimeter that utilizes low thermal mass (i.e., low Phi factor) test cells, such as the Phi-TEC II, means that very little of the heat produced during the reaction and runaway is consumed in warming up the test cell. This enables the conditions of a large-scale plant reaction to be emulated, with the maximum temperature attained (Tend) and the rate of pressure increase representative of what would be expected to occur during a plant-scale incident. The kinetic data obtained can be used to calculate the adiabatic temperature rise of the runaway (ΔTad,d) and the time to maximum rate of decomposition (TMRd). Unlike adiabatic calorimeters with a high Phi factor, the reaction rate is not suppressed by the high thermal mass of the test cell. So little to no data manipulation is required to compensate for heat losses to the test cell.
Comparing Reaction Scenarios
The Phi-TEC II also enables different reaction scenarios to be simulated and compared. In the example introduced earlier, the worst-case scenario that would release the most energy is the reaction of equimolar amounts of the alcohol and anhydride.
Temperature
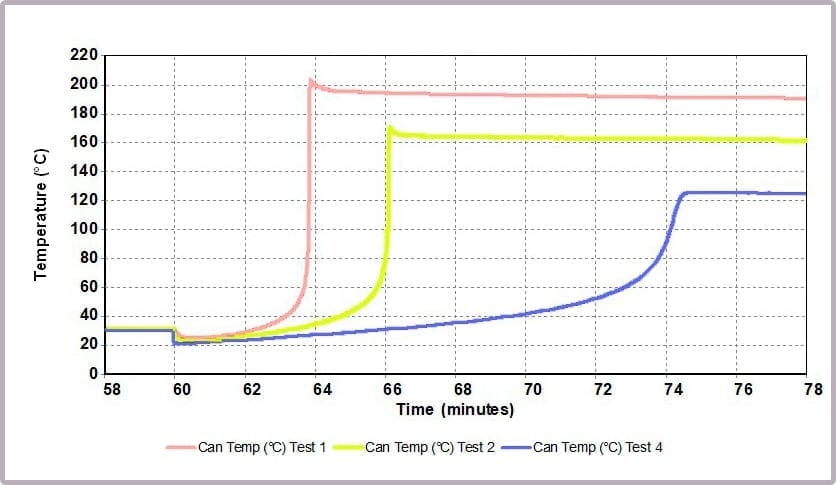
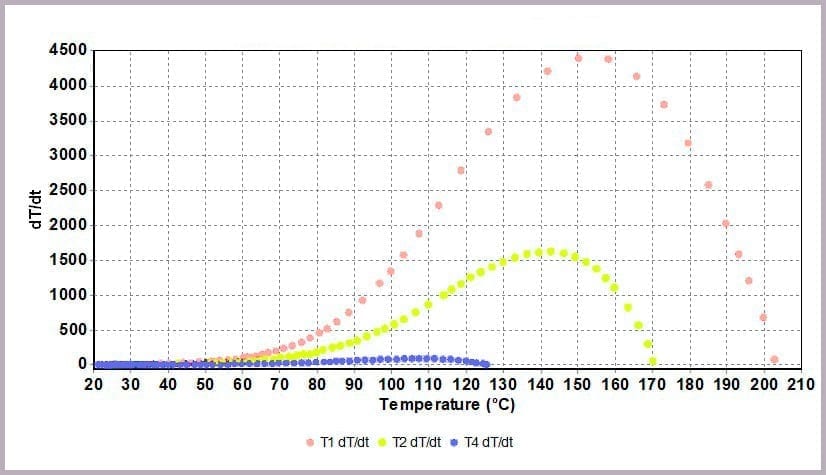
Comparing the temperature results, it can be seen from the plot in Figure 11a that halving the amount of anhydride reduces the maximum temperature from over 200⁰C to 170⁰C and the time taken to reach that point. This change is further highlighted by considering the plot of the rate of temperature against temperature (the plot in Figure 11b), where the maximum rate reached drops from 4000⁰C/min to just over 1600⁰C/min when the amount of anhydride is halved.
These reactions were carried out using a Phi-TEC II on a low Phi-factor test cell and under adiabatic conditions. The parameters describing the exothermic event (MTSR, TMR, ΔTad, Tend) are highly representative of what would be expected to be observed in a plant-scale incident.
Pressure
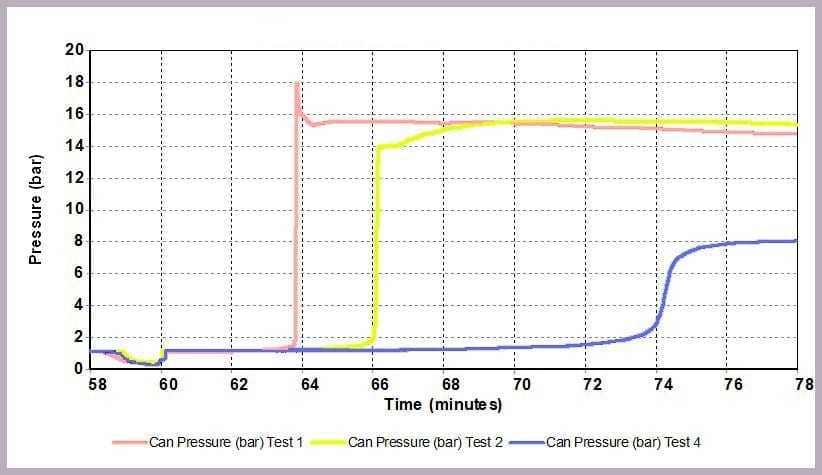
As Figure 12 illustrates, a similar effect can be seen with the pressure data: when the level of anhydride is reduced, the maximum pressure reached is reduced, and the rate of pressure increase is slowed. In this example, the lower pressure rise directly results from the lower temperature.
Safety Considerations
The data illustrates that for the worst-case scenario, it barely takes 3 minutes (after the chemicals come together) for the reaction to runaway to completion, and in the process:
- reaching a temperature of approximately 200⁰C
- a pressure of 16 bar
- with the rate of temperature increase peaking at 4000⁰C/min.
In contrast, by reducing the anhydride amount to a quarter that of the alcohol, the incident takes 12 minutes to reach the worst conditions, which are now reduced to:
- a maximum temperature of 125⁰C
- a maximum pressure of 8 bar
- a peak rate of temperature change of 150⁰C/min.
This is because the higher proportional levels of alcohol act as a thermal diluent, leading to the lower final temperature, lower final pressure, and a lower rate of increase in temperature and pressure observed.
Subsequently, this information enables the correct combination of reactor type and protection features to be selected, which could include the following:
- Emergency & evaporative cooling
- Quenching
- Controlled depressurization
- Vent sizing
Thus, directly comparing the impact of different operating scenarios enables the necessary safety controls to be selected upfront for the manufacturing plant.
Solutions
The Phi-TEC II is a more advanced type of adiabatic calorimeter, which supports using low Phi factor test cells. This capability means that very little of the heat produced during a reaction or thermal runaway is consumed in warming the test cell. As a result, the runaway rate is not tempered.
The measured rate of pressure increase and final temperature (Tend), along with the calculated Time to Maximum Rate (TMR) and adiabatic temperature rise (ΔTad), are representative of what would be expected to occur during a manufacturing scale incident. Thus, the Phi-TEC II enables the hazards to be thoroughly evaluated and explored, facilitating their mitigation before scale-up.
Phi-TEC II | Bench-top, low phi-factor, adiabatic calorimeter
Home › Products › Process Safety › Phi-TEC IIPhi-TEC IIFully customi...
